- Орфанные заболевания >
- Болезнь Фабри >
- Лизосомные болезни накопления
- Болезнь Фабри
- Историческая справка
- Генетика болезни Фабри: Наследование
- Молекулярная генетика и синтез α-галактозидазы А
- Мутации в гене GLA
- Гено-фенотипические корреляции
- Синтез альфа-галактозидазы А
- Клинические проявления болезни Фабри
- Периферическая нервная система: Нейропатические боли
- Гипогидроз / гипергидроз
- Центральная нервная система
- Почечная патология
- Сердечные нарушения
- Кожные изменения
- Офтальмологические нарушения
- Нарушения слуха
- Желудочно-кишечные расстройства
- Дыхательные нарушения
- Качество жизни пациентов с БФ
- Другие проявления болезни Фабри
- Клиническая картина болезнь Фабри с поражением одной функциональной системы или органа
- Клинические проявления болезни Фабри: Заключение
- Клинические проявления БФ у женщин и детей
- Клинические проявления БФ у детей
- Клинические проявления БФ у лиц женского пола
- Диагностика болезни Фабри
- Введение
- Дифференциальная диагностика ангиокератом
- Болевой синдром
- Нервная система
- Почечная патология
- Сердечно-сосудистая система
- Офтальмологические нарушения
- Желудочно-кишечный тракт
- Качество жизни
- Биохимическая и генетическая диагностика БФ
- Пренатальная диагностика БФ
- Генетическое консультирование
- Диспансеризация пациентов с БФ
- Диагностика болезни Фабри: Заключение
- Лечение болезни Фабри
- Симптоматическое лечение: Лечение болевого синдрома
- Почечная патология при БФ
- Сердечно-сосудистые нарушения
- Ангиокератомы
- Желудочно-кишечные расстройства
- Фермент-заместительная терапия
- Генотерапия
- Фармакологические шапероны
- Ограничение синтеза субстрата
- Лечение болезни Фабри: Заключение
- Важность гликозилирования при разработке фермент-заместительной терапии
- Перенос α-галактозидазы А
- Гликозилирование
- Гликозилирование α-галактозидазы А
- Заключение
- Клинический эффект ФЗТ
- Периферическая нервная система: Нейронопатические боли
- Гипогидроз / гипергидроз
- Центральная нервная система
- Почечная патология
- Сердечно-сосудистая система
- Кожные проявления
- Орган зрения
- Орган слуха
- Желудочно-кишечный тракт
- Качество жизни
- Клинический эффект ФЗТ: Заключение
- Список литературы
- Болезнь Фабри >
Статьи и публикации
Лизосомные болезни накопления
Лизосомные болезни накопления (ЛБН) – группа наследственных заболеваний, включающая более 45 различных нозологических форм, каждая из которых связана с нарушением определенной функции лизосом. Эти нарушения приводят к накоплению внутри клеток макромолекул, которые в норме подвергаются гидролизу в лизосомах. Нарушения на различных стадиях одного метаболического пути приводит к разным заболеваниям (рис. 1). ЛБН разделяют на группы в зависимости от типа накапливаемых макромолекул (мукополисахаридозы, гликопротеинозы, сфинголипидозы). Клинические проявления этих нарушений сходны и включают патологию скелета, увеличение размера внутренних органов (гепатоспленомегалию), поражение центральной нервной системы, изменение черт лица (огрубление), изменение структуры волос.
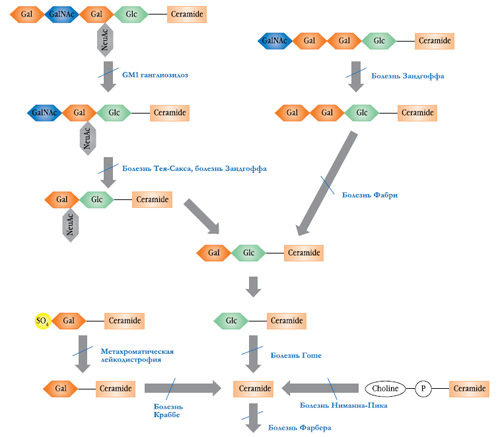
Рис. 1. Разные формы лизосомных болезней накопления связаны с дефектами в определенном метаболическим пути.
Опубликовано с разрешения Beck & Ries (2001). Gal – галактоза; GalNAc – N-ацетил-D-галактозамин; Glc – глюкоза; NeuAc – N-ацетилнейраминовая кислота
Большинство ЛБН наследуются по аутосомно- рецессивному типу, за исключением болезни Фабри, синдрома Хантера, синдрома Данон, которые наследуются по Х-сцепленному типу. Суммарная частота ЛБН составляет 1 : 4000 – 1 : 8000 живых новорожденных (Meikle et al., 1999; Pinto et al., 2004; Poorthuis et al., 1999). Каждое из заболеваний встречается крайне редко, с частотой от 1: 57 000 (болезнь Гоше), 1 : 42 000 000 (сиалидоз) (Meikle et al., 1999). В разных популяциях частота ЛБН может различаться, так исследования, проведенные в Голландии, показали, что частота болезни Гоше составляет 1 : 86 200, а частота болезни Помпе 1 : 50 000 живых новорожденных (Poorthuis et al., 1999).
Большинство ЛБН относятся к числу прогрессирующих наследственных заболеваний, сопровождаются ранней инвалидизацией и приводят к преждевременной смерти. Однако наблюдаются существенные различия в сроках дебюта болезни и скорости прогрессирования не только между отдельными ЛБН, но даже в пределах одной нозологической формы. Лечение ЛБН заключается прежде всего в симптоматической терапии. Трансплантация гемопоэтических клеток применяется для некоторых заболеваний, однако, как показывают клинические испытания, исход трансплантации очень варьирует, а для некоторых форм не приводит к значительным клиническим улучшениям (Hoogerbrugge & Valero, 1998). Другие терапевтические подходы – лечение фармакологическими шаперонами, генотерапия только разрабатываются. На сегодняшний день одним из самых успешных подходов к лечению ЛБН является фермент-заместительная терапия (ФЗТ). ФЗТ применяется для лечения мукополисахаридоза II типа, мукополисахаридоза I типа, болезнях Гоше, Фабри и Помпе. На первых стадиях клинических испытаний находится терапия и для ряда других ЛБН.