- Орфанные заболевания >
- Болезнь Фабри >
- Лизосомные болезни накопления
- Болезнь Фабри
- Историческая справка
- Генетика болезни Фабри: Наследование
- Молекулярная генетика и синтез α-галактозидазы А
- Мутации в гене GLA
- Гено-фенотипические корреляции
- Синтез альфа-галактозидазы А
- Клинические проявления болезни Фабри
- Периферическая нервная система: Нейропатические боли
- Гипогидроз / гипергидроз
- Центральная нервная система
- Почечная патология
- Сердечные нарушения
- Кожные изменения
- Офтальмологические нарушения
- Нарушения слуха
- Желудочно-кишечные расстройства
- Дыхательные нарушения
- Качество жизни пациентов с БФ
- Другие проявления болезни Фабри
- Клиническая картина болезнь Фабри с поражением одной функциональной системы или органа
- Клинические проявления болезни Фабри: Заключение
- Клинические проявления БФ у женщин и детей
- Клинические проявления БФ у детей
- Клинические проявления БФ у лиц женского пола
- Диагностика болезни Фабри
- Введение
- Дифференциальная диагностика ангиокератом
- Болевой синдром
- Нервная система
- Почечная патология
- Сердечно-сосудистая система
- Офтальмологические нарушения
- Желудочно-кишечный тракт
- Качество жизни
- Биохимическая и генетическая диагностика БФ
- Пренатальная диагностика БФ
- Генетическое консультирование
- Диспансеризация пациентов с БФ
- Диагностика болезни Фабри: Заключение
- Лечение болезни Фабри
- Симптоматическое лечение: Лечение болевого синдрома
- Почечная патология при БФ
- Сердечно-сосудистые нарушения
- Ангиокератомы
- Желудочно-кишечные расстройства
- Фермент-заместительная терапия
- Генотерапия
- Фармакологические шапероны
- Ограничение синтеза субстрата
- Лечение болезни Фабри: Заключение
- Важность гликозилирования при разработке фермент-заместительной терапии
- Перенос α-галактозидазы А
- Гликозилирование
- Гликозилирование α-галактозидазы А
- Заключение
- Клинический эффект ФЗТ
- Периферическая нервная система: Нейронопатические боли
- Гипогидроз / гипергидроз
- Центральная нервная система
- Почечная патология
- Сердечно-сосудистая система
- Кожные проявления
- Орган зрения
- Орган слуха
- Желудочно-кишечный тракт
- Качество жизни
- Клинический эффект ФЗТ: Заключение
- Список литературы
- Болезнь Фабри >
Статьи и публикации
Историческая справка
Болезнь Фабри (БФ) впервые была описана независимо двумя дерматологами из Германии и Англии (рис. 3). Джон Фабри (1860-1930) родился в Германии и изучал медицину в Боне. Он работал в Королевской клинике дерматовенерологических заболеваний (Konigliche Klinik fur Haut und Geschlechtskrankheiten) в Бонне 1886-1889. С апреля 1889 он возглавил отделение дерматологии в Дормунде.
Вильям Андерсон (1842-1900) родился в Лондоне и начал академическую карьеру учеником школы искусств. Он очень хотел заниматься медициной и поступил на работу в госпиталь Св. Томаса (St. Thomas hospital). Некоторое время он работал помощником хирурга, потом переехал в Японию и возглавил военно-морской медицинский колледж (Japanese Naval Medical College) в Токио. В 1880 году он вернулся в госпиталь Св. Томаса, в котором и продолжал свою медицинскую карьеру.
В 1898 году Фабри описал 13-летнего мальчика с нодулярной пурпурой, у которого впоследствии развилась альбуминурия. Он классифицировал данный случай как один из вариантов диффузной ангиокератомы (рис. 4). В этом же году Андерсон
описал 39-летнего мужчину с ангиокератомой, протеинурией, деформациями пальцев рук, варикозным расширением вен и лимфоотеком (рис. 5). Андерсон предположил наличие мультисистемного заболевания (Anderson, 1898). Более распространенное название болезни – болезнь Фабри, другой вариант названия – болезнь Андерсона-Фабри.

Рис. 3. (a) Джон Фабри (1860-1930) и его публикация, посвященная болезни Фабри; (б) Вильям Андерсон (1842-1900) и его статья
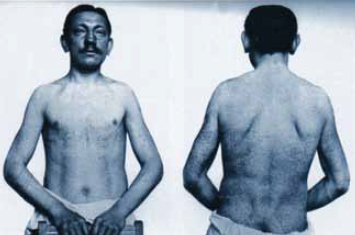
Рис. 4. Диффузная ангиокератома туловища (Angiokeratoma corporis diffusum.) Фотография сделана в 1915 году, прогрессирование кожных изменений с 1898 г. (Fabry, 1916)
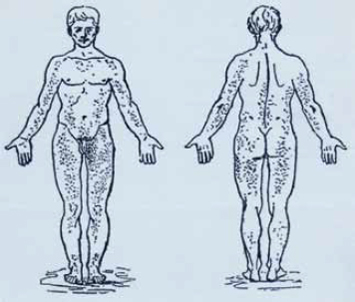
Рис. 5. Диффузная ангиокератома туловища (Angiokeratoma corporis diffusum.) Рисунок из работы Андерсона (1898)
Дальнейшие публикации, посвященные этому заболеванию, в начале ХХ века также включали описание гипогидроза, акропарастезий и офтальмологических нарушений. В 1947 году Pompen et al. описали аномальные вакуоли в кровеносных сосудах и предположили, что заболевание относится к группе болезней накопления. В 1950 г. была определена природа накапливаемых соединений. Было показано, что они представляют собой жиры. В 1953 г. впервые подтвердили диагноз БФ при проведении биопсии кожи (Hornbostel & Scriba, 1953; Scriba, 1950). Затем Opitz с соавторами (Opitz and colleagues, 1965) в 1965 году описали тип наследования заболевания, хотя ранее Wise et al. (1962) также отмечали семейный характер болезни. Заболевание было отнесено к группе сфинголипидозов после определения Sweeley и Klionsky структуры накапливаемых жиров (Sweeley & Klionsky, 1963). В 1967 году Brady с соавторами открыли первичный биохимический дефект – недостаточность церамидтригексоздазы (названной позднее α-галактозидазой А). И в 1970 году Kint было показано значительное снижение активности этого фермента в тканях пациентов с БФ (Brady et al., 1967; Kint, 1970). В дальнейшем стало известно, что недостаточность α-галактозидазы А является причиной БФ, а недостаточность α-галактозидазы В – болезни Шиндлера (Dean et al., 1977; Schindler et al., 1989; Schram et al., 1977). В 1989 году ген α-галактзидазы А был секвенирован, что дало возможность с помощью генно-инженерных методов синтезировать данный фермент in vitro (Kornreich et al., 1989). Это привело к созданию фермент-заместительной терапии для БФ.