Синдромам Гурлер-Шейе и Шейе (I-V и V типы мукополисахаридозов) свойственно более легкое течение болезни, и у страдающих больных, как правило, нормальный интеллект. В основе развития этих трех синдромов лежит недостаточность одного и того же лизосомного фермента – a-L-иду- ронидазы.
Синдром Хантера (II тип мукополисахаридоза) отличается от синдрома Гурлер типом наследования (болеют, как правило, только мальчики) и отсутствием помутнения роговицы. Выделение легкой формы синдрома Хантера обусловлено нормальным интеллектом этих больных.
III тип мукополисахаридоза (синдромы Санфилиппо A, B, C и D) характеризуют нормальные показатели физического развития, меньшая тяжесть скелетных и органных изменений и грубое снижение интеллекта.
Детей с VI типом мукополисахаридоза (синдром Марото-Лами) отличают карликовый рост и высокий коэффициент интеллектуального развития (IQ).
Синдрому Слая (VII тип мукополисахаридоза) свойственны «гурлер-подобный» фенотип и грубая задержка психоречевого развития.
Для IV типа мукополисахаридоза (синдром Моркио A и B) характерны карликовость, килевидная деформация грудной клетки, увеличение объема и вальгусная установка коленных суставов, гиперподвижность межфаланговых и контрактуры крупных суставов, диффузная мышечная гипотония, помутнение роговицы и тугоухость. Интеллектуальное развитие больных не страдает.
В практическом плане все типы мукополисахаридозов удобнее делить на две группы – «гурлер-подобный» (рис. 1а, в) и «моркио-подобный» фенотипы (рис. 1б). Последний включает синдром Моркио А и В, а остальные 12 объединет «гурлер-подобный» фенотип.
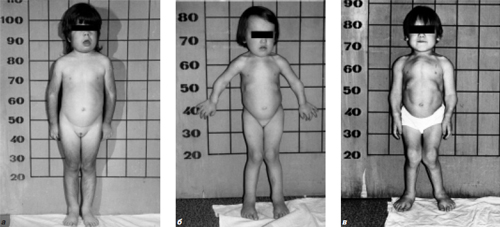
Рис. 1. а – ребенок 8 лет с VI типом мукополисахаридоза (синдром Марото-Лами); б – девочка 4 лет с синдромом Моркио А; в – девочка 15 лет с синдромом Гурлер-Шейе.
