Выявление мутации, ответственной за заболевание, позволило провести ДНК-диагностику у других членов семьи с целью выявления носительства у сестры больного. Выявление носительства мутаций у родственников больных с болезнью Фабри имеет практическое значение. В настоящее время известно, что гетерозиготные носительницы мутаций в гене X-Gal могут иметь клинические признаки заболевания от минимально выраженных до значительных, и также нуждаются в динамическом наблюдении и лечении [7]. В данной семье 3 женщины II.1, III.3, IV.1 (рис. 4, А) являются носительницами мутации, исследование носительства мутации у ребенка (IV.3) не проводилось.
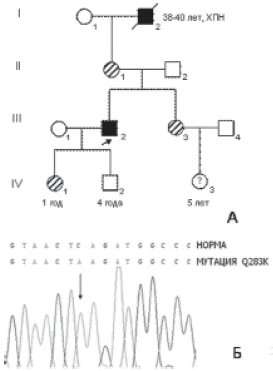
Рисунок 4. Генетическое исследование.
А – родословная семьи П. Римскими цифрами отмечены номера поколений, арабскими – номер человека в поколении. Стрелкой отмечен пробанд (пациент П.). Темными символами отмечены больные члены семьи, штрихованными – женщины – носитель¬ницы мутации p.Glu238Lys; Б – фрагмент прямого секвенирования последовательности экзона 6 гена X-Gal пациента П. Стрелкой отмечено место мутации. Вверху приведена расшифровка выявленной нуклеотидной последовательности, для сравнения выше приведен нормальный фрагмент последовательности гена X-Gal.
Больному проводилась следующая терапия: арифонретард 1,5 мг/сут, омез 40 мг/сут, де-нол. По завершении противоязвенного курса повторно проведена эзофагогастродуоденоскопия, при этом эрозий не выявлено, назначена терапия тромбо-АССом 100 мг/сут. После выписки из клиники отметил постепенное снижение переносимости физических нагрузок, во время которых стали беспокоить слабость, головокружение, сохранялась выраженная брадикардия. В РНЦХ РАМН им. акад. Б.В. Петровского пациенту имплантирован постоянный электрокардиостимулятор в режиме VVIR, после чего самочувствие больного заметно улучшилось. Проконсультирован кардиохирургом – показаний к хирургическому лечению митральной недостаточности и проведению пластики левого предсердия на момент консультации нет.
Патогенетической терапией болезни Фабри является пожизненная ферментная заместительная терапия [5]. В этом году в России были зарегистрированы два препарата, представляющие собой рекомбинантные белки: α-галактозидаза и ß-галактозидаза. В настоящее время решается вопрос о предоставлении пациенту П. ферментной заместительной терапии.
Заключение
Болезнь Фабри – наследственное мультисистемное заболевание, обусловленное мутациями в гене X-Gal, кодирующем лизосомный фермент α-галактозидазу. Снижение активности фермента ведет к прогрессирующему накоплению глоботриазоилкерамида (GB-3) и родственных гликосфинголипидов в лизосомах клеток эндотелия сосудов, нервных, миокардиальных, всех типах ренальных клеток[4]. Заболевание наследуется по Х-сцепленному рецессивному типу, при этом женщины – носительницы мутаций также могут иметь клинические признаки заболевания. По данным европейских скрининговых исследований, распространенность заболевания составляет около 1:40 000 населения [2], однако в некоторых клинических группах его частота значительно выше. По данным разных авторов, 3-12% пациентов с необъяснимой гипертрофией левого желудочка (чаще всего концентрической), около 2% больных с ишемическими инсультами [6] и 0,5% больных с хронической почечной недостаточностью страдают этим заболеванием [2, 4, 6]. В настоящее время частота выявления болезни Фабри остается низкой. Хотя клинические признаки заболевания проявляются еще в детском возрасте, заболевание долгое время остается нераспознанным. Средний возраст больных в момент постановки правильного диагноза составляет около 30 лет. Приведенный клинический пример выявления болезни Фабри у пациента П. в возрасте 32 лет служит примером такой диагностики. Болезнь Фабри необходимо исключать, если у пациента наблюдается сочетание 3 следующих признаков или более:
1. Гипертрофическая необструктивная кардиомиопатия.
2. Микроальбуминурия, протеинурия или хроническая почечная недостаточность неясной этиологии.
3. Ранние инсульты (до 55 лет).
4. Ангиокератомы, особенно в нижней трети живота, паховой и ягодичной областях.
5. Артериальная гипертензия.
6. Акропарестезии (ощущение жжения или покалывания в кончиках пальцев кистей и стоп, особенно на холоде или в тепле), плохая переносимость жары и холода, частые ожоги или обморожения.
7. Гипогидроз (сниженное потоотделение).
8. Нарушения зрения и/или слуха.
9. Наличие вышеперечисленных заболеваний среди родственников, особенно мужского пола.
