Из анамнеза известно, что ребенок от III беременности, протекавшей на фоне анемии, носительства золотистого стафилококка, хронической внутриутробной гипоксии (I беременность – медицинский аборт, II – антенатальная гибель плода в сроке 38 недель, внутриутробная асфиксия). Роды II срочные, маловодие, околоплодные воды зеленые. Масса тела при рождении 3090 г, длина – 47 см, окружность головы 35 см, груди 34 см, оценка по шкале Апгар 7-8 баллов.
С первых суток отмечались срыгивания после каждого кормления, на 2-е сутки – резкое ухудшение состояния, подъем температуры тела до 38,5°С, беспокойство, тонические судороги на фоне гипогликемии.
По результатам неонатального скрининга обнаружена гиперфенилаланинемия: 2,75 - 17,3 - 6,06 мг % (норма 1-2 мг %).
Для уточнения наследственной природы заболевания проводилось исследование крови и мочи в лаборатории наследственных болезней обмена ГУ Медико-генетического центра РАМН г. Москва:
1) активность галактозо-1-фосфат уридилтрансферазы в пределах нормы – 4,98 ед/мин/г гемоглобина (норма 4,4-15), исключающая классическую галактоземию;
2) методом высокоэффективной жидкостной хроматографии установлено превышение концентрации сукцинилацетона в моче почти в 10 раз – 197 мМ/М креатинина (норма 0-20), что является патогномоничным признаком тирозинемии 1-го типа;
3) методом тандемной масс-спектрометрии выявлено умеренное повышение уровня тирозина – 264 мкМ/л (норма 11-200), а также повышение метионина – 229 мкМ/л (норма 6-65), являющиеся диагностическими маркерами тирозинемии 1-го типа.
На основании комплекса полученных данных на 15-й день пребывания в стационаре был подтвержден диагноз: тирозинемия 1-го типа, острая форма. Для лечения ребенка решался вопрос о закупке специальной аминокислотной смеси Тирозидон (Нутриция). Но течение заболевания имело исключительно тяжелый и быстропрогрессирующий характер с летальным исходом.
Данные патоморфологического и гистологического исследования печени свидетельствовали о неспецифическом циррозе печени с атрофией гепатоцитов, холестазом и имбибицией желчью гепатоцитов (рис. 3), изменениях в почках в виде зернистой дистрофии эпителия извитых канальцев вплоть до некроза в сочетании с микрокальцинозом канальцев и ишемией клубочков.
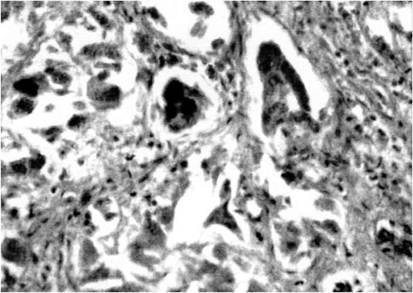
Рисунок 3. Гистологический препарат печени больной К. с тирозинемией 1-го типа.
Наше наблюдение показывает, что необходимым условием успешной и быстрой диагностики данной группы наследственных заболеваний является тесное взаимодействие педиатров и генетиков для совместного планирования тактики обследования в каждом конкретном случае, так как эти заболевания не обладают достаточно характерной клинической симптоматикой и для их верификации требуется применение специальных лабораторных методов исследования. Чрезвычайно важно неотложное решение вопроса о специфическом лечении больного, хотя не при всех формах оно дает положительный клинический эффект.
ЛИТЕРАТУРА
1. Burlina AB, Bonafe L, Zacchello F. Seminars in Perinatology. 1999; 23,2: 162-173.
2. Краснополъская К.Д. Наследственные болезни обмена веществ. Справочное пособие для врачей. М.: РОО «Центр социальной адаптации и реабилитации детей «Фохат», 2005: 364.
3. Наследственные нарушения нервно-психического развития детей. Под ред. П.А. Темина, Л.З. Казанцевой. М.: Медицина, 2001: 432.
4. Педиатрия. Руководство. Книга 2. Болезни плода и новорожденного, врожденные нарушения обмена веществ: Пер. с англ. Под ред. Р.Е. Бермана, В.К. Вогана. 2-е изд. М.: Медицина, 1991: 528.
5. Lindstedt S, Holme E, Lock EA et al. Lancet. 1992; 340: 813-817.
